Médicament injectable : le parcours d’un combattant
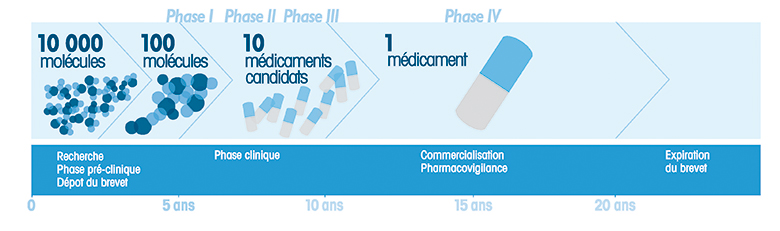
De la recherche sur les molécules à la commercialisation du médicament injectable, 12 à 13 années de développement sont nécessaires. La « gestation » du médicament en poche, elle, connaît les mêmes quatre phases que les médicaments classiques, plus quelques spécificités. Éclairage sur un itinéraire semé d’embûches.
Une fois la recherche exploratoire et les tests précliniques sur les molécules achevés, les essais cliniques de la phase I débutent sur des volontaires sains. Ils permettent de déterminer la dose maximale tolérée chez l’homme et son mode d’administration. La phase II évalue la relation dose-effets, définie la posologie et détecte les effets indésirables à court terme. Elle marque également le début du développement de l’emballage primaire. Pour être compatible avec le produit et assurer sa parfaite stabilité, le matériau de la future poche doit respecter de nombreux paramètres. On cherche à éviter toute interaction avec le produit qui constituerait une menace pour la qualité du médicament injectable et donc pour la sécurité du patient.
Point clef : la matière première de la poche. Elle est soumise à une étude des extractibles et des relargables, puis à une évaluation de la toxicité et des risques. A ce stade, chez Technoflex, la R&D vérifie aussi que la compatibilité entre la poche et le bec de remplissage est optimale (diamètre, remplissage aseptique ou non,…).
D’autres facteurs, hormis ceux directement liés à la composition du médicament qui entrent dans sa formulation, peuvent affecter la stabilité du produit en contact avec le contenant. C’est le cas de la température ambiante et de l’humidité. L’étude de stabilité qui est menée doit aussi tenir compte des basses températures ainsi que des cycles de congélation-décongélation, notamment pour les biotechnologies et les dérivés sanguins. Pour certaines préparations, il est également indispensable de considérer les effets d’une exposition à la lumière.
La phase III pourrait être appelée la phase de « l’essai comparatif ». On confronte les propriétés du médicament avec un placebo ou un médicament existant. Seule la preuve d’un rapport bénéfice-risque acceptable permet l’obtention de l’AMM (Autorisation de Mise sur le Marché). A ce stade des essais, l’emballage primaire du médicament injectable est abouti : polypropylène, EVA ou PVC selon la nature du contenu. L’ultime phase débute lorsque le médicament est commercialisé. C’est celle de la pharmacovigilance. On continue ainsi d’approfondir la connaissance du médicament dans les conditions réelles d’utilisation.
Développer des médicaments injectables de qualité et efficaces est l’objectif de la recherche thérapeutique. C’est un parcours complexe et de longue haleine. L’expertise de tous les acteurs du développement est nécessaire à l’émergence de traitements innovants pour la santé des patients.
- Les extractibles désignent les composés qui peuvent être extraits des matières plastiques par des solvants de propriété physico-chimiques différentes dans des conditions agressives.
- Les relargables font référence aux composés qui peuvent être libérés par les matières plastiques dans le produit pharmaceutique dans des conditions normales d’utilisation.
Sylvie Ponlot


















